
Facilities
Biophysical techniques
Wide-ranging techniques to answer an array of technical questions, such as discovering interactions between molecules, investigating conformational changes, and determining binding kinetics.
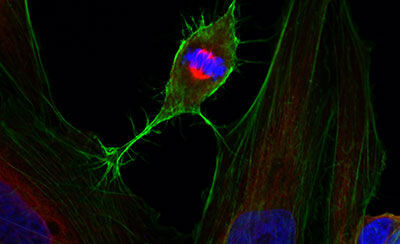
Cellular techniques
Diverse equipment for producing high quality recombinant proteins and imaging cells and tissues, including wide-field, confocal or super-resolution fluorescence systems, electron microscopy, flow cytometric analysis, and support with sample preparation.
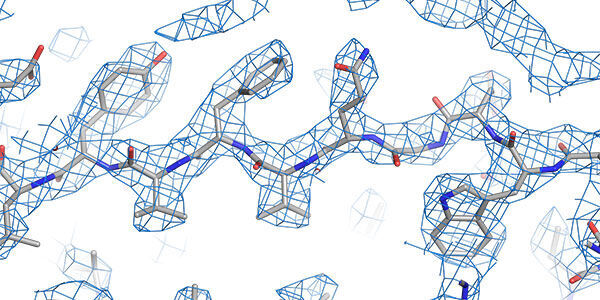
Structural techniques
World leading state-of-the-art facilities for investigating the structure and conformation of biomolecules down to the atomic scale.

Technical Services
Our world-leading research facilities are supported by our dedicated Technical Services team, who work across all areas of our research.